
化学と材料科学の分野は、MetaのFundamental AI Research(FAIR)チームによる最新のリリース、FAIRChemモデルにより大きな変革を迎えています。このブログでは、FAIRChemとは何か、その主要コンポーネントであるUniversal Model for Atoms(UMA)やOpen Molecules 2025(OMol25)データセット、そして研究者やイノベーターにとってなぜ重要なのかを紹介します。
FAIRChemとは?
FAIRChemは、MetaのFAIR Chemistryチームが開発した、化学および材料科学のためのAI駆動ソリューションを推進する集中型リポジトリです。データセット、事前学習済みモデル、デモ、アプリケーションを統一フレームワークで提供し、研究者が分子や材料の発見にAIを活用しやすくします。MITライセンスの下で公開されており、GitHubやHugging Faceを通じてアクセス可能で、オープンサイエンスとコラボレーションを促進します。
以前はOpen Catalyst Projectとして知られていたFAIRChemは、バージョン2のリリースで大きく進化しました。なお、FAIRChem V2はV1との互換性がなく、従来のモデルやコードは使用できません。
基盤モデル
Universal Model for Atoms (UMA)
UMA(Universal Model for Atoms)は、FAIRChemの中心的なモデルです。分子や材料の原子レベル表現を統一的に扱えるよう設計されており、多様なタスクに応用できます。UMAは、エネルギー推定、力の予測、反応経路解析といった量子化学的な問題に幅広く対応可能です。
UMAの特徴:
- 汎用性:単一モデルで複数の分子・材料シナリオに対応
- スケーラビリティ:大規模データセットを活用した事前学習
- 拡張性:Hugging Face Hubを通じて追加のファインチューニングが容易
これにより、研究者は一からモデルを構築せずに、高度な予測タスクに直接取り組むことができます。
データセット
Open Catalyst 2020 (OC20) ~ 2022(OC22)
Open Catalyst データセットは、触媒表面に分子が吸着したときの構造やエネルギーを予測するための大規模データセットです。数百万件に及ぶ吸着構造と力・エネルギーラベルを収録しており、触媒設計の加速を目的としています。表面科学やエネルギー変換研究において標準的なベンチマークとなっています。 初版はOC20で現在最新のバージョンはOC22になります。
OMAT (Open Materials)
結晶や固体材料を対象としたデータセットです。結晶構造やバンドギャップ、安定性といった物性データを収録しており、新材料探索のための重要なリソースとなります。半導体設計やエネルギー材料研究に活用可能です。
OMOL (Open Molecules)
分子を対象とした大規模データセットです。
数千万分子を網羅し、量子化学計算に基づく高品質ラベルを含みます。分子物性予測や薬物探索、材料設計の基盤となります。
ODAC (Open Direct air capture)
金属有機構造体(MOFs)に特化したデータセットです。
ガス吸着や分離、触媒応用などに広く用いられるMOF材料をカバーし、構造探索や機能予測を支援します。
OMC (Open Molecular Crystals)
分子結晶を対象としたデータセットです。
有機結晶の安定性や結晶構造予測に利用でき、電子材料や光学材料の設計に重要です。
FAIRChemの重要性
FAIRChemは、AIモデルの訓練に必要な包括的で高精度なデータセットの不足という、分子および材料科学の重大な課題に対処します。従来のDFT計算は計算コストが高く、実験的手法は時間がかかります。FAIRChemのデータセットとUMAのような事前学習済みモデルは、高スループットのスクリーニングやシミュレーションを前例のない規模で可能にし、触媒、材料、分子の新たな発見を加速します。
例えば、FAIRChemを活用することで、研究者は以下のようなことが可能です。
- 水素貯蔵などの再生可能エネルギー用途の触媒を最適化する。
- 直接空気回収や次世代コンピューティングハードウェアのための新素材を設計する。
- 生体分子や反応性システムの分子動力学シミュレーションを高精度で実行する。
FAIRChemのインストール
1. FAIRChemを使用するには、Hugging FaceアカウントとUMAモデルリポジトリへのアクセスが必要です。UMAモデルにアクセスするには、連絡先情報を共有することに同意する必要があります。
UMAモデルにアクセスするために連絡先情報共有に同意
出典:https://huggingface.co/facebook/UMA
2. 仮想環境を作るのをお勧めします。今回はcondaで作ります。
conda create -n farichem_env
3. pipやuvを使ってインストールします。今回はpipでインスタールします。
pip install ase
pip install fairchem-core
Web DEMO
UMAモデルではウェブ上での利用も可能になっています。
UMA ストラクチャー
出典:https://huggingface.co/spaces/facebook/fairchem_uma_demo
UMAモデル インプット画面
出典:https://huggingface.co/spaces/facebook/fairchem_uma_demo
 UMAモデル セッティング画面
UMAモデル セッティング画面
出典:https://huggingface.co/spaces/facebook/fairchem_uma_demo
 UMAモデル セッティング画面
UMAモデル セッティング画面
出典:https://huggingface.co/spaces/facebook/fairchem_uma_demo
FAIRChemの分子動力学シミュレーション(MD)
今回は例として、tert-butanolのMDを行ってみます。以下はMDを機械学習ベースのポテンシャル、機械学習ポテンシャル (MLIP, Machine Learning Interatomic Potential)で実行するコード です。
1. 必要なライブラリをインストール
from ase.io import write
from ase.io import Trajectory
from ase.visualize import view
from ase.md.langevin import Langevin
from ase import units
from fairchem.core import pretrained_mlip, FAIRChemCalculator
from rdkit import Chem
from rdkit.Chem import AllChem
from ase import Atoms
2. 化学構造は一般的なファイルタイプをサポートする ase.Atoms オブジェクトを作成することでロードできます。本家のサイトでは"H2O"を指定していましたが、ASE の molecule() には “H2O” などのごく基本的な分子のみがプリセットされています。tert-butanolはプリセットに無いため、そのまま molecule("t-BuOH") のようには呼び出せません。そのためRDKit で SMILES → 3D 座標生成 → ASE へ渡すという手法を紹介します。
# 1. tert-butanolのSMILESを作成
smiles = "CC(C)(C)O"
rdmol = Chem.MolFromSmiles(smiles)
rdmol = Chem.AddHs(rdmol)
# 2. 3D座標を生成
AllChem.EmbedMolecule(rdmol, AllChem.ETKDG())
AllChem.UFFOptimizeMolecule(rdmol)
# 3. 原子の位置と記号を抽出
conf = rdmol.GetConformer()
positions = np.array([list(conf.GetAtomPosition(i)) for i in range(rdmol.GetNumAtoms())])
symbols = [atom.GetSymbol() for atom in rdmol.GetAtoms()]
# 4. ASE Atomsオブジェクトを作成
mol = Atoms(symbols=symbols, positions=positions)
# 5. 真空パディングを追加
mol.center(vacuum=5.0)
mol.pbc = (False, False, False)
# 6. .trajに保存
write("tBuOH_pre_md.traj", mol)
3. 作成したtert-butanolを可視化
traj = Trajectory('tBuOH_pre_md.traj')
view(traj)
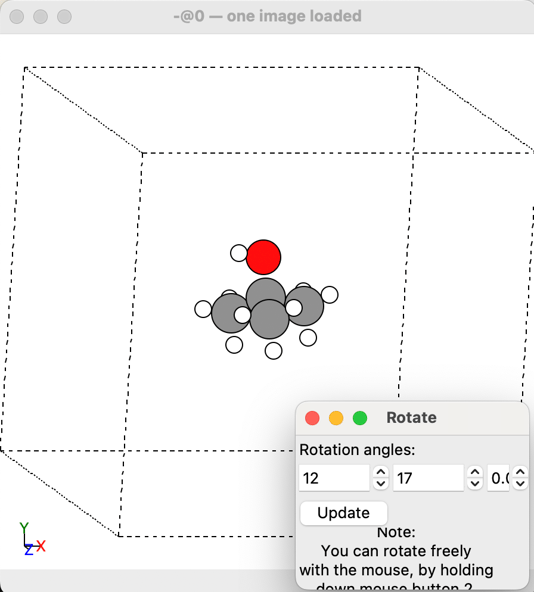
tert-butanolの3D図
4. MDシミュレーションを実行
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1", device="cpu")
calc = FAIRChemCalculator(predictor, task_name="omol")
# atoms = molecule("H2O")
mol.calc = calc
dyn = Langevin(
mol,
timestep=0.1 * units.fs,
temperature_K=400,
friction=0.001 / units.fs,
)
trajectory = Trajectory("tBUOH_post_md.traj", "w", mol)
dyn.attach(trajectory.write, interval=1)
dyn.run(steps=1000)
5. MDシミュレーション結果を可視化
traj_md = Trajectory('tBUOH_post_md.traj')
view(traj_md)
tert-butanolの熱振動シミュレーション
感想
先日紹介したIBMのMIのファウンデーションモデル FM4M は、MetaのFAIRChemにおける UMA と同様に、材料科学のための基盤モデルを目指すものです。ただし、FM4M は SMILES を中心とした分子表現の学習に基盤を置いているのに対し、UMA は原子座標や結晶構造を直接扱うという点でアプローチが異なります。そのため、IBM のモデルは分子特性予測や創薬研究に強みを持ち、FAIRChem は触媒や無機材料、分子結晶、MOFs までを包括的にカバーし、材料科学やエネルギー分野での活用に直結しています。両者は競合関係ではなく、分子中心と原子・材料中心という補完的なアプローチであり、研究の目的に応じて使い分けることが最も効果的だといえるでしょう。
また、MIの発展においては依然として「データ不足」が大きな課題となっています。シミュレーションデータを基盤として構築されたこれらのファウンデーションモデルは、その制約を補い、膨大な化学空間をこれまでにない速度と精度で探索するための不可欠なツールになると考えています。
参考資料
FAIRChem Repository
https://github.com/facebookresearch/fairchem?tab=readme-ov-fileMeta release news
https://ai.meta.com/blog/meta-fair-science-new-open-source-releases/
FAIRChem UMA demo
https://huggingface.co/spaces/facebook/fairchem_uma_demoUMA Repository
https://huggingface.co/facebook/UMA
Profile
EAGLYS MIリサーチャー&エンジニア
温庭立/Uen Tinnlea
東京大学 バイオエンジニアリング専攻 博士課程修了。データサイエンティストとしての経歴を経て、EAGLYSでMI分野の研究・開発に従事する。2024年には早稲田大学 創造理工学部講義 マテリアルズインフォマティクス 講師も。日本語能力検定 N1
